









Клиническими испытаниями мы обязаны талидомиду
В настоящее время необходимость проведения качественных КИ препаратов не вызывает вопросов. Требования регуляторных органов к организации КИ постоянно совершенствуются. Как следствие, усложняется дизайн исследований, растет число пациентов-участников и врачей-исследователей, увеличиваются сроки наблюдения. Это приводит к росту затрат разработчика и значительно задерживает выход на рынок новых препаратов. В то же время такой тщательный контроль позволяет избежать тяжелых последствий, возможных при широком применении малоизученных лекарств. Но так было не всегда. До середины XX века, когда во всем мире происходило становление фармацевтической промышленности, масштабные КИ еще не были обязательным условием для вывода препаратов в широкое обращение. Все изменилось после «талидомидовой трагедии», которая привела к кардинальному пересмотру требований к изучению безопасности новых лекарств.
Так, в частности, было введено обязательное изучение в эксперименте на животных тератогенности всех лекарств. Во многих странах были разработаны требования к проведению КИ с участием людей перед выводом новых лекарств на рынок. Принята поправка к американскому «Закону о лекарственных средствах» (Drug Amendment Act) 1962 года, которая диктует необходимость проведения производителями предрегистрационных исследований для доказательства не только безопасности, но и эффективности нового препарата, и вскоре такие исследования стали обычной практикой в большинстве стран мира. Также «талидомидовая трагедия» привела к развитию системы мониторинга побочных действий лекарств после их выхода на рынок.
Регуляторы всех стран, объединяйтесь!
Первые руководства и законы, разработанные в разных государствах для регуляции КИ, неизбежно различались между собой. В 1980-х годах страны Европы начали создавать единое экономическое пространство, которое позднее легло в основу Евросоюза. В рамках данного процесса необходимо было сформировать общий рынок лекарственных средств. Принятие единых требований к регистрации и обращению лекарств позволило бы избежать дублирования исследований и избыточных бюрократических процедур. Конкретные планы по унификации были названы в 1989 году на Международной конференции регуляторных органов в сфере обращения лекарственных средств, организованной ВОЗ (WHO International Conference of Drug Regulatory Authorities). В результате в 1990 году представители не только стран Европы, но и США и Японии создали Международный совет по гармонизации технических требований к фармацевтическим препаратам для человека (International Council for Harmonisation, ICH).
ICH стал организацией, объединившей регуляторные органы разных стран и фармацевтическую промышленность, чтобы совместными усилиями создать универсальные международные научные и технические требования к разработке лекарств, проведению КИ, производству и обращению препаратов. В настоящее время участниками ICH являются страны Евросоюза, США, Япония, Канада, Бразилия, Китай и другие государства. Россия и страны ЕАЭС выступают в качестве наблюдателей.
ICH разработал множество руководств, которые поделены на четыре категории: качество, безопасность, эффективность и мультидисциплинарные руководства. Ознакомиться с ними можно на официальном сайте организации. Основная часть руководств ICH, посвященных эффективности и безопасности лекарственных средств, имеет непосредственное отношение к проведению КИ. Они рассматривают этические и научные вопросы, статистические методы и дизайн исследований. Особое внимание уделяется изучению токсичности лекарств.
Первая версия GCP R1 была опубликована в 1996 году. В ней были описаны права и обязанности всех участников КИ, включая исследователей, мониторов (контролеров), представителей спонсора и этических комитетов, а также требования к документированию исследования. В 2016 году вышло руководство GCP R2 – дополненная версия, освещающая методы более эффективного проведения исследований и вопросы электронного документооборота.
Протокол планируемого КИ – цели, дизайн, число участников – формируется исходя из стратегии фармацевтической компании с учетом регуляторных требований стран, где планируются продажи разрабатываемого лекарства. С целью одновременной регистрации в нескольких государствах организуются международные многоцентровые КИ. В таких исследованиях изучение препарата происходит одновременно в нескольких странах по единому протоколу.
Как упоминалось в нашей первой статье, посвященной КИ, при создании документов, регулирующих сферу КИ в России и ЕАЭС, за основу были взяты международные рекомендации, разработанные ICH. Однако пока что в нашей стране официально лишь часть руководств соответствует требованиям ICH. Это различие осложняет вывод российских препаратов на международные рынки, так как отечественные исследования зачастую не соответствуют международным требованиям.
Клинические исследования оригинальных лекарств и дженериков: в чем разница
Существует огромная разница между КИ оригинальных препаратов и дженериков (лекарственных препаратов, идентичных запатентованным по действующему веществу).
Для регистрации большинства дженериков, за исключением отдельных лекарственных форм, достаточно проведения одного исследования биоэквивалентности, обычно на небольшой группе здоровых добровольцев (30–100 человек). В ходе такого исследования сравнивается биодоступность (скорость и степень всасывания) действующего вещества. Подтверждение биоэквивалентности позволяет экстраполировать результаты доклинических и клинических исследований оригинального препарата на дженерик.
Напомним, что согласно Федеральному закону № 61 «Об обращении лекарственных средств», не требуется проведение исследований биоэквивалентности для регистрации отдельных видов дженериков.
Три фазы исследования: зачем и почему
В соответствии с регуляторными требованиями, чтобы доказать эффективность и безопасность оригинальных лекарств, необходимо провести полный цикл КИ.
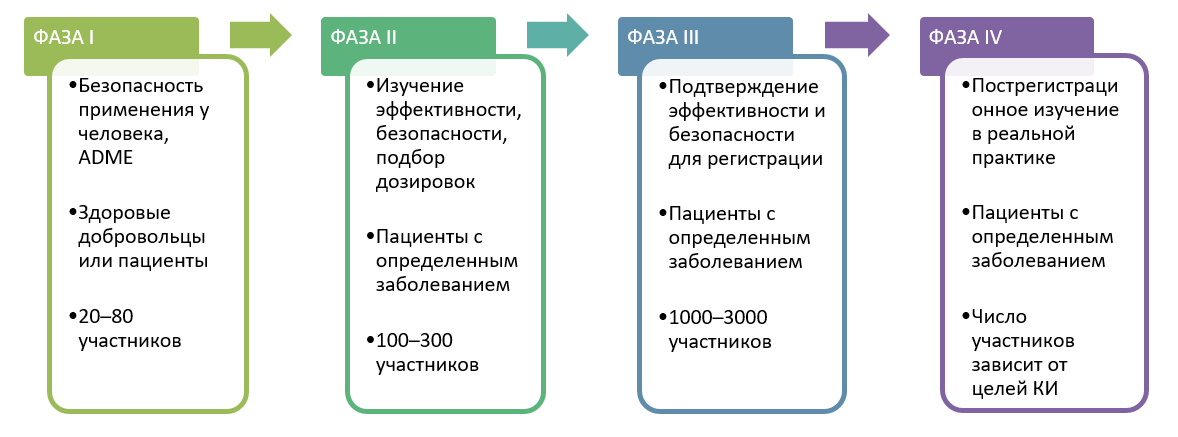
Число участников первых испытаний составляет десятки человек и увеличивается до сотен и тысяч в последующих КИ, необходимых для доказательства эффективности и безопасности препарата. Конкретное число пациентов зависит от дизайна исследования и рассчитывается статистически исходя из величины клинически значимого эффекта, количества сравниваемых групп, вариабельности изучаемых параметров, а также допустимого уровня погрешностей (ошибки первого и второго рода).
Цель фазы I – оценка безопасности и переносимости активного вещества при первом применении у человека. Такие испытания проводят на небольшом числе здоровых добровольцев (20–80), как правило молодых мужчин. Помимо обязательной страховки, участники получают финансовую компенсацию за то, что подвергают свое здоровье риску. Если препарат заведомо вреден для здорового человека (например, цитостатик, предназначенный для лечения онкологических заболеваний), то в исследованиях этой фазы принимают участие пациенты с конкретным заболеванием. Допустимую безопасную дозу препарата и путь введения обычно устанавливают в предварительных экспериментах на животных и корректируют по результатам исследований. В КИ I фазы удается изучить, как препарат всасывается, распределяется, метаболизируется, выводится (ADME), вызывает ли нежелательные реакции. Проводится также предварительная оценка эффективности.
Положительные результаты КИ I фазы позволяют продолжить изучение лекарства. В ходе КИ II фазы оценивают эффективность и безопасность применения лекарства у небольших групп пациентов с определенным заболеванием (100–300 человек). Изучается зависимость эффекта от дозы и продолжительности приема. Исследуется безопасность многократного применения препарата по схеме лечения. При проведении КИ II фазы подбирается оптимальная доза препарата, оказывающая выраженный терапевтический эффект с минимальным количеством побочных реакций. В дизайне КИ II фазы обязательно наличие группы сравнения (это контролируемое исследование). Пациенты группы сравнения проходят стандартную, хорошо изученную терапию данного заболевания или получают плацебо.
Основная задача исследований II фазы – получение сведений о новом лекарстве, достаточных для принятия решения о том, целесообразно ли продолжать его изучение, поскольку исследования III фазы являются наиболее затратными.
Клинические исследования III фазы называют также регистрационными: их успешное завершение позволяет зарегистрировать препарат и вывести его на рынок. Для того чтобы результаты КИ были максимально достоверными, III фазу проводят, как правило, в виде рандомизированного контролируемого двойного слепого многоцентрового исследования на больших группах пациентов (сотни или тысячи человек). Так же как и в исследованиях II фазы, обязательно сравнение со стандартной терапией или плацебо. Рандомизация означает, что пациенты распределяются по группам случайным образом. А двойной слепой метод предполагает, что ни пациент, ни врач-исследователь не знают, какой именно препарат получает пациент. Чем больше пациентов учавствует в КИ, тем больше вероятность выявить побочные явления, связанные с приемом препарата. Дополнительные исследования фазы III необходимы также для тщательного изучения безопасности и эффективности лекарств у различных групп пациентов, а также в сочетании с другими лекарствами. Исследования фазы III являются наиболее длительными, по сравнению с более ранними фазами, особенно при изучении препаратов для лечения хронических заболеваний. В них участвуют сотни и тысячи пациентов.
После успешного завершения исследования III фазы и подтверждения эффективности и безопасности препарата его досье может быть направлено в уполномоченный орган для государственной регистрации.
Далеко не все КИ дают желаемый результат. Согласно статистическому исследованию, опубликованному в 2021 году, вероятность успешного завершения КИ I фазы составляет около 50 %, II фазы – 30 %, III фазы – 60 %. С учетом этих вероятностей шанс успешного завершения всех трех фаз КИ составляет лишь 10 %. Это означает, что лишь один из десяти экспериментальных препаратов будет зарегистрирован. При этом наименьший шанс на успех имеют препараты для лечения онкологических заболеваний – всего 5 %.
Для участия в КИ I–III фаз отбирают участников, отвечающих весьма строгим критериям включения – обычно исключают пожилых, с сопутствующими заболеваниями, беременных, детей, курящих и злоупотребляющих алкоголем и т. д. Правильность приема лекарства контролирует врач – невозможно пропустить прием или принять лишнюю таблетку. В обычной жизни все обстоит несколько иначе, поэтому организуются пострегистрационные исследования, они же исследования IV фазы. Они помогают выявить редкие побочные эффекты и взаимодействие с другими лекарствами, изучить влияние длительного приема на организм человека.
Тяжелые побочные эффекты, обнаруженные в ходе исследований IV фазы и пострегистрационного мониторинга безопасности, серьезно меняют соотношение польза/риск. В этом случае применение препарата у определенных групп пациентов может быть ограничено или вовсе запрещено. Например, церивастатин был изъят из обращения фирмой Bayer в 2001 году из-за смертельных случаев рабдомиолиза, которые были выявлены в ходе его широкого применения во многих странах.
Другой пример – лоркасерин, препарат для подавления аппетита и длительной терапии ожирения. Он был одобрен в США в 2012 году при условии проведения пострегистрационного рандомизированного двойного слепого плацебо-контролируемого КИ с участием 12 тыс. пациентов. При анализе данных исследования, длившегося более 5 лет, был выявлен повышенный риск развития рака у людей, принимавших лоркасерин. В результате он был отозван с рынка в 2020 году.
Часто пострегистрационные исследования носят наблюдательный характер, являются неинтервенционными (т. е. препарат применяется в рутинной практике в соответствии с одобренной инструкцией без дополнительного вмешательства исследователей). Пострегистрационные исследования способствуют маркетинговому продвижению препаратов, повышению осведомленности врачей об их эффектах.
Заключение
Существует множество видов КИ, и все они проводятся для того, чтобы обеспечить людей наиболее эффективными и безопасными лекарствами. Основной международный стандарт проведения КИ – «Надлежащая клиническая практика» – одно из руководств, созданных ICH. Участники ICH – страны Евросоюза, США, Япония, Канада, Бразилия, Китай и другие государства. Россия и страны ЕАЭС являются наблюдателями. У нас официально принята лишь часть руководств, соответствующих требованиям ICH, что создает трудности для вывода российских препаратов на международные рынки лекарств.
Рандомизированные КИ – «золотой стандарт» для определения безопасности и эффективности новых лекарств. Они классифицируются по фазам в зависимости от дизайна, целей и регистрационного статуса препарата. Однако даже крупные исследования III фазы не могут предоставить полные данные о безопасности препарата, поскольку число пациентов в этих исследованиях ограничено. Размер выборки позволяет доказать эффективность лекарства, но зачастую не дает возможности обнаружить редкие нежелательные реакции. Поэтому необходимо проведение исследований IV фазы, непрерывный сбор и анализ сообщений о побочных эффектах при приеме препаратов в рутинной практике.
В экстренной ситуации новые лекарственные препараты получают регистрационное удостоверение по ускоренной процедуре при условии обязательного дальнейшего проведения пострегистрационного исследования безопасности. Например, КИ вакцины против новой коронавирусной инфекции «Гам-Ковид-Вак» (Sputnik V) с участием более 31 тысячи человек началось в августе 2020 года, практически сразу после ее государственной регистрации в России.
Читайте также:
Клинические исследования новых лекарств: как получить разрешение на проведение испытаний

В 1957 году в Германии, а затем еще в 45 странах препарат талидомид назначался как седативное средство, а также для устранения утренней тошноты у беременных женщин. Согласно требованиям того времени, для подтверждения безопасности лекарства было достаточно провести эксперименты на животных. Талидомид был признан безопасным и, благодаря активной рекламе, быстро стал популярен. Хотя вскоре начали появляться сообщения о поражении периферических нервов на фоне приема препарата, он продолжал лидировать на рынке, и производители упорно отрицали эту связь. Позже было замечено увеличение числа новорожденных с тяжелыми пороками развития конечностей (фокомелией) и других органов. Их матери принимали талидомид в период беременности. Всего пострадало более 10 тыс. детей. В 1961 году, когда связь наблюдаемых осложнений с приемом талидомида была доказана, его применение было запрещено.
Оригинальными препаратами называют новые лекарства, активное вещество которых было впервые синтезировано и ранее не применялось у человека. Они, как правило, защищены патентами на определенный срок. По истечении патентной защиты любые фармацевтические компании имеют право создавать дженерики, которые содержат то же самое действующее вещество, но могут отличаться по составу вспомогательных ингредиентов, консервантов и т. д. Кроме того, может отличаться и технология производства, например степень очистки действующего вещества.
- предназначенных для парентерального введения и представляющих собой водные растворы;
- представляющих собой растворы для перорального применения;
- произведенных в форме порошков или лиофилизатов для приготовления растворов;
- являющихся газами;
- являющихся ушными или глазными лекарственными препаратами, произведенными в форме водных растворов;
- предназначенных для местного применения и приготовленных в форме водных растворов;
- представляющих собой водные растворы для использования в форме ингаляций с помощью небулайзера или в качестве назальных спреев, применяемых с помощью сходных устройств.





